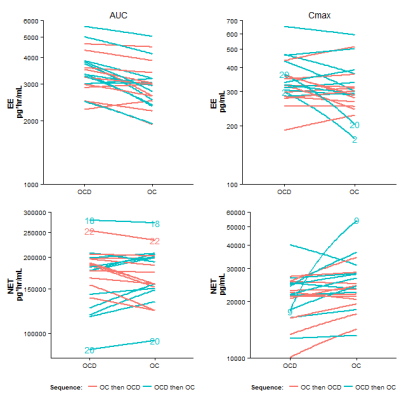
This post is actually a continuation of the previous post, and is motivated by this article that discusses the graphics and statistical analysis for a two treatment, two period, two sequence (2x2x2) crossover drug interaction study of a new treatment versus the standard. Whereas the previous post was devoted to implementing some of the graphics presented in the article, in this post we try to recreate the statistical analysis calculations for the data from the drug interaction study. The statistical analysis is implemented with R.
Dataset:
The drug interaction study dataset can be obtained from ocdrug.dat.txt and a brief description of the dataset is at ocdrug.txt. We first save the files to a local directory and then read the data into R dataframe and assign appropriate column labels:
ocdrug <- read.table(paste(workdir,"ocdrug.dat.txt",sep=""),sep="")
## “workdir” is the name of the variable containing the directory name where the data file is stored
colnames(ocdrug) <- c("ID","Seq","Period","Tmnt","EE_AUC","EE_Cmax","NET_AUC","NET_Cmax")
## Give nice names to the treatments (OCD and OC) and the treatment sequence
ocdrug$Seq <- factor(ifelse(ocdrug$Seq == 1,"OCD-OC","OC-OCD"))
ocdrug$Tmnt <- factor(ifelse(ocdrug$Tmnt == 0,"OC","OCD"), levels = c("OC", "OCD"))
Then log transform the PK parameters:
ocdrug$logEE_AUC <- log(ocdrug$EE_AUC)
ocdrug$logEE_Cmax <- log(ocdrug$EE_Cmax)
ocdrug$logNET_AUC <- log(ocdrug$NET_AUC)
ocdrug$logNET_Cmax <- log(ocdrug$NET_Cmax)
Analysis based on Normal Theory – Linear Mixed Effects Model
We illustrate the normal theory based mixed effects model for the analysis of EE AUC. The analysis for EE Cmax and for NET AUC and Cmax are similar. We use the recommended R package nlme for developing the linear mixed effects models. The models include terms for sequence, period and treatment.
require(nlme)
options(contrasts = c(factor = "contr.treatment", ordered = "contr.poly"))
lme_EE_AUC <- lme(logEE_AUC ~ Seq+Period+Tmnt, random=(~1|ID), data=ocdrug)
From the fitted models, we can calculate the point estimates and confidence intervals for the geometric mean ratios for AUC and Cmax for EE and NET. This is illustrated for EE AUC:
tTable_EE_AUC <- summary(lme_EE_AUC)$tTable["TmntOCD",]
logEst <- tTable_EE_AUC[1]
se <- tTable_EE_AUC[2]
df <- tTable_EE_AUC[3]
t <- qt(.95,df)</pre>
## Estimate of geometric mean ratio
gmRatio_EE_AUC <- exp(logEst)
## 90% lower and upper confidence limits
LCL_EE_AUC <- exp(logEst-t*se); UCL_EE_AUC <- exp(logEst+t*se)
The calculations for Cmax and for NET are similar. We can bind all the results from normal theory model into a dataframe:
norm.out <- data.frame(type=rep("Normal Theory",4),
cmpd=c(rep("EE",2), rep("NET",2)),
PK=rep(c("AUC","Cmax"),2),
ratio=c(gmRatio_EE_AUC, gmRatio_EE_Cmax, gmRatio_NET_AUC, gmRatio_NET_Cmax),
LCL=c(LCL_EE_AUC, LCL_EE_Cmax, LCL_NET_AUC, LCL_NET_Cmax),
UCL=c(UCL_EE_AUC, UCL_EE_Cmax, UCL_NET_AUC, UCL_NET_Cmax))
rownames(norm.out) <- paste(norm.out$cmpd,norm.out$PK,sep="_")
cat("\nSummary of normal theory based analysis\n")
cat("---------------------------------------\n")
print(round(norm.out[,4:6],2))
As discussed in the article, the diagnostic plots to assess the appropriateness of the normal theory based linear model, i.e., normal probability plots, scatter plots of observed vs. fitted values, and scatter plots of studentized residuals vs. fitted values, show some deviations from assumptions, at least for Cmax. Hence, a distribution-free or non-parametric analysis may not be unreasonable.
Distribution-free Analysis
A distribution free analysis for bioequivalence is using the two sample Hodges-Lehmann point estimator and the two sample Moses confidence interval. Difference in treatment effects are estimated as the average difference in the two treatment sequence groups with respect to the within-subject period difference in the log AUC (or log Cmax) values. But we must first calculate these within-subject period differences. Transforming the dataset to a ‘wide’ format may be useful for the calculation of these within-subject differences:
ocdrug.wide <- reshape(ocdrug, v.names=c("EE_AUC", "EE_Cmax", "NET_AUC", "NET_Cmax",
"logEE_AUC", "logEE_Cmax", "logNET_AUC", "logNET_Cmax", "Tmnt"), timevar="Period",
idvar="ID", direction="wide", new.row.names=NULL)
ocdrug.wide$logEE_AUC_PeriodDiff <- with(ocdrug.wide, logEE_AUC.1 - logEE_AUC.2)
Similarly, the within-subject differences can be calculated for EE Cmax and NET AUC and Cmax. The two sample Hodges-Lehmann point estimator and the two sample Moses confidence interval can be calculated from these within subject differences using the the pairwiseCI function in the R-package pairwiseCI with the method=HL.diff option. Also, as specified in the article, we need to divide the estimates and CI bounds by 2 because the difference is taken twice (sequence and period).
npEst_EE_AUC <- unlist(as.data.frame(pairwiseCI(logEE_AUC_PeriodDiff ~ Seq, data=ocdrug.wide,
method="HL.diff", conf.level=0.90)))
npRatio_EE_AUC <- as.numeric(exp(npEst_EE_AUC[1]/2))
npLCL_EE_AUC <- as.numeric(exp(npEst_EE_AUC[2]/2))
npUCL_EE_AUC <- as.numeric(exp(npEst_EE_AUC[3]/2))
Similarly, the estimates and confidence intervals can be obtained for EE Cmax and NET AUC and Cmax. We bind all the results obtained from distribution free theory analysis into a dataframe:
np.out <- data.frame(type=rep("Distribution Free",4),
cmpd=c(rep("EE",2), rep("NET",2)),
PK=rep(c("AUC","Cmax"),2),
ratio=c(npRatio_EE_AUC, npRatio_EE_Cmax, npRatio_NET_AUC, npRatio_NET_Cmax),
LCL=c(npLCL_EE_AUC, npLCL_EE_Cmax, npLCL_NET_AUC, npLCL_NET_Cmax),
UCL=c(npUCL_EE_AUC, npUCL_EE_Cmax, npUCL_NET_AUC, npUCL_NET_Cmax))
rownames(np.out) <- paste(np.out$cmpd,np.out$PK,sep="_")
cat("\nSummary of distribution-free analysis\n")
cat("-------------------------------------\n")
print(round(np.out[,4:6],2))
Finally, we try to recreate figure 13 that graphically compares the results of the normal theory and distribution free analyses:
# We collect the quantities to plot in a dataframe and include a dummy x-variable
plotIt <- rbind(np.out,norm.out)
plotIt$x <- c(1,3,5,7,1.5,3.5,5.5,7.5)
# Then plot
png(filename = paste(workdir,"plotCIs.png",sep=""), width = 640, height = 640)
par(font.lab=2, font.axis=2, las=1, font=2, cex=1.6, cex.lab=0.8, cex.axis=0.8)
col=c(rep("magenta",4), rep("lightseagreen",4))
lty=c(rep(2,4),rep(1,4))
pch=c(rep(17,4), rep(16,4))
plot(ratio ~ x, data=plotIt, type="p", pch=pch, col=col, axes=FALSE,
xlim=c(0.5,8), ylim=c(0.7,1.35), xlab="", ylab="Ratio(OCD/OC)")
arrows(plotIt$x, plotIt$LCL, plotIt$x, plotIt$UCL,
code=3, length = 0.08, angle = 90, lwd=3, col=col, lty=lty)
axis(side=2, at=c(0.7,0.8,1, 1.25))
text(x=c(1,3,5,7), y=rep(0.7,4), pos=4, labels=rep(c("AUC","Cmax"),2), cex=0.8)
text(x=c(2,6), y=rep(0.65,2), pos=4, labels=c("EE","NET"), xpd=NA, cex=0.8)
abline(h=c(0.8,1,1.25),lty=3)
legend("top",c(paste("Distribution-Free","\t\t"),"Normal Theory"),
col=unique(col), lty=unique(lty), pch=unique(pch), horiz=TRUE, bty="n", cex=0.8, lwd=2)
dev.off()
Results
Summary of normal theory based analysis
---------------------------------------
ratio LCL UCL
EE_AUC 1.16 1.11 1.23
EE_Cmax 1.10 1.01 1.19
NET_AUC 1.01 0.97 1.04
NET_Cmax 0.86 0.78 0.95
Summary of distribution-free analysis
-------------------------------------
ratio LCL UCL
EE_AUC 1.16 1.11 1.24
EE_Cmax 1.08 0.99 1.20
NET_AUC 1.00 0.97 1.04
NET_Cmax 0.90 0.82 0.96

These results appear in tables 2-3 and figure 13 of the article.











{kind=link}
{kind=link}